Generating Hi-C matrices with fanc¶
This part of the documentation will focus primarily on fanc auto - the most versatile
command in the FAN-C toolkit. Its main goal is to convert any input to binned Hi-C matrices.
The following schematic will give you an overview of what file types fanc auto can handle
and how they are processed downstream.
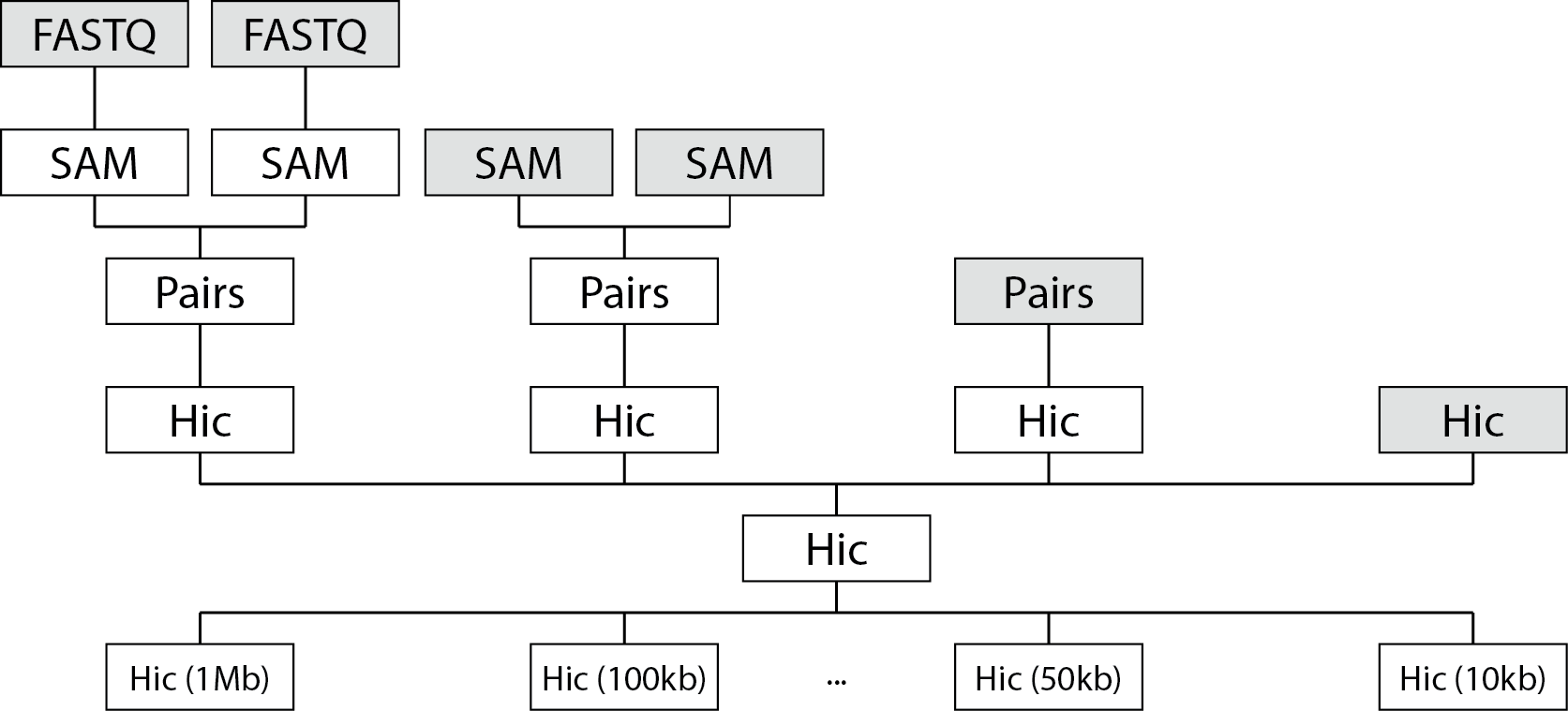
fanc auto will map reads in FASTQ (or gzipped FASTQ) files to a reference genome, generating
SAM/BAM files. SAM/BAM files with paired-end reads will be automatically sorted and mate pairs
will be matched to generate Pairs files. Pairs files will be converted into fragment-level Hic
objects. Multiple fragment-level Hic objects will be merged into a single Hi-C object. Finally,
the fragment-level Hic object will be binned at various bin sizes.
Internally, fanc auto constructs its Hi-C processing pipeline from more specialised fanc
commands. When describing the different pipeline steps and how you can control them below, we
will also reference the specialised command that is used to build each step of the pipeline.
Parameters¶
Each pipeline step in fanc auto is controlled by a specific set of parameters in fanc auto.
Some of these are mandatory for specific types of input, others are optional and affect, for example,
the processing and filtering of objects in the pipeline.
Here is the help output for fanc auto:
usage: fanc auto [-h] [-g GENOME] [-r RESTRICTION_ENZYME] [-i GENOME_INDEX]
[-n BASENAME] [-s STEP_SIZE] [-b BIN_SIZES [BIN_SIZES ...]]
[-t THREADS]
[--max-restriction-site-distance MAX_RESTRICTION_SITE_DISTANCE]
[--fanc-parallel] [--split-fastq] [--memory-map] [--ice]
[--norm-method NORM_METHOD] [-q QUALITY_CUTOFF]
[--iterative-quality-cutoff ITERATIVE_QUALITY_CUTOFF]
[--le-inward-cutoff INWARD_CUTOFF]
[--le-outward-cutoff OUTWARD_CUTOFF] [--auto-le-cutoff]
[-tmp] [--iterative] [--no-sam-sort] [--restore-coverage]
[--split-ligation-junction] [--no-filter-pairs] [--no-hic]
[--run-with RUN_WITH] [--job-prefix JOB_PREFIX]
[--grid-startup-commands GRID_STARTUP_COMMANDS]
[--grid-cleanup-commands GRID_CLEANUP_COMMANDS] [-f]
[--no-sambamba USE_SAMBAMBA]
input [input ...] output_folder
Positional Arguments¶
| input | Input files. fanc will try to guess the file type by its extension. |
| output_folder | Output folder. All output files and folders will be generated under this directory. |
Named Arguments¶
| -g, --genome | Genome for the Hi-C object.Path to region-based file (BED, GFF, …) containing the non-overlapping regions to be used for Hi-C object construction. Typically restriction-enzyme fragments. Alternatively: Path to genome file (FASTA, folder with FASTA, hdf5 file), which will be used in conjunction with the type of restriction enzyme (-r) to calculate fragments directly. |
| -r, --restriction-enzyme | |
| Restriction enzyme name. Used for in silico digestion of genomic sequences and splitting of reads at Hi-C ligation junctions. (e.g. HindIII, case-sensitive). Separate multiple enzymes with ‘,’. Restriction names can be any supported by Biopython, which obtains data from REBASE (http://rebase.neb.com/rebase/rebase.html). | |
| -i, --genome-index | |
| Bowtie 2 or BWA genome index. Only required when passing FASTQ files as input. | |
| -n, --basename | Basename for output files. If not provided, will be guessed based on input file names. |
| -s, --step-size | |
| Step size for iterative mapping. Default: 3 | |
| -b, --bin-sizes | |
| Bin sizes for Hi-C matrix generation. Default: 5mb, 2mb, 1mb, 500kb, 250kb, 100kb, 50kb, 25kb, 10kb, 5kb. | |
| -t, --threads | Maximum number of threads. The number provided here will not be exceeded by analysis steps running alone or in parallel. Default: 1 |
| --max-restriction-site-distance | |
| Insert / ligation fragment sizes are inferred from the sum of distances of both reads to the nearest restriction sites. Fragment larger than this value are filtered from the Pairs object. The default value of 10000 only removes extremely large fragments, and is thus considered conservative.Default: 10000 | |
| --fanc-parallel | |
| Use FAN-C parallelisation, which launches multiple mapper jobs. This may be faster in some cases than relying on the internal parallelisation of the mapper, but has potentially high disk I/O and memory usage. | |
| --split-fastq | Split fastq files into chunks of 10M reads. Reads will be merged again on the SAM level. Splitting and merging bypasses the -tmp flag. This option reduces disk usage in tmp, in case the system has a small tmp partition. |
| --memory-map | Map Bowtie2 index to memory. Recommended if running on medium-memory systems and using many parallel threads). |
| --ice | DEPRECATED. Correct Hi-C matrices using ICE instead of Knight-Ruiz matrix balancing. Slower, but much more memory-friendly. |
| --norm-method | Normalisation method. Options are: KR (default) = Knight-Ruiz matrix balancing (Fast, accurate, but memory-intensive); ICE = ICE matrix balancing (more CPU-intensive, but also more memory-efficient); VC = vanilla coverage (a single round of ICE balancing); VC-SQRT = vanilla coverage square root (reduces overcorrection compared to VC) |
| -q, --quality-cutoff | |
| Cutoff for the minimum mapping quality of a read. For numbers larger than 1, will filter on MAPQ. If a number between 0 and 1 is provided, will filter on the AS tag instead of mapping quality (only BWA). The quality cutoff is then interpreted as the fraction of bases that have to be matched for any given read. Only applies to SAM/BAM input!. Default is not to filter on mapping quality. | |
| --iterative-quality-cutoff | |
| MAPQ cutoff for mapped reads. Only applies when iterative mapping is enabled: if a mapped read has MAPQ below this cutoff,it will be sent to another iteration in an attempt to find a higher quality alignment. Default is 3 for BWA and 30 for Bowtie2. | |
| --le-inward-cutoff | |
| Ligation error inward cutoff. Default: no ligation error filtering. | |
| --le-outward-cutoff | |
| Ligation error outward cutoff. Default: no ligation error filtering. | |
| --auto-le-cutoff | |
| Automatically determine ligation error cutoffs. Use with caution, this setting has a tendency to choose large cutoffs, removing many pairs close to the diagonal. | |
| -tmp, --work-in-tmp | |
| Work in temporary directory. Copies input files and generates output files in a temporary directory. Files will be moved to their intended destination once an analysis step finishes. Reduces network I/O if using remote file systems. | |
| --iterative | Map reads iteratively. Can improve mappability, especially with low-quality reads. Reads are initially trimmed to 25bp and mapped to the reference genome. If no unique mapping location is found, the read is extended by 3bp and the process is repeated until the full length of the read is reached or a unique mapping location is found. |
| --no-sam-sort | Do not sort SAM/BAM files. Sorted files are required for the pair generating step. Only omit this if you are supplying presorted (by read name) SAM/BAM files. |
| --restore-coverage | |
| Restore coverage to the original total number of reads. Otherwise matrix entries will be contact probabilities. Only available for KR matrix balancing. | |
| --split-ligation-junction | |
| Split reads at predicted ligation junction before mapping. Requires the -r argument. | |
| --no-filter-pairs | |
| Do not filter read pairs. By default, the following filters are applied: self-ligations, PCR duplicates,restriction distance (>10kb) | |
| --no-hic | Do not process pairs into Hi-C maps (stop after read pairing step). |
| --run-with | Choose how to run the commands in fanc auto. Options: ‘parallel’ (default): Run fanc commands on local machine, use multiprocessing parallelisation. ‘sge’: Submit fanc commands to a Sun/Oracle Grid Engine cluster. ‘slurm’: Submit fanc commands to a Slurm cluster. ‘test’: Do not run fanc commands but print all commands and their dependencies to stdout for review. |
| --job-prefix | Job Prefix for SGE and Slurm. Works with ‘–run-with sge’ and –run-with slurm. Default: ‘fanc_<6 random letters>_’ |
| --grid-startup-commands | |
| Path to a file with BASH commands that are executed before every FAN-C command that is run on a grid engine / cluster. This could, for example, include environment-specific settings, such as activation of a Python virtualenv. | |
| --grid-cleanup-commands | |
| Path to a file with BASH commands that are executed after every FAN-C command that is run on a grid engine / cluster. Use this to clean the file system or environment set up with –grid-startup-commands. | |
| -f, --force-overwrite | |
| Force overwriting of existing files. Otherwise you will be prompted before files are overwritten. | |
| --no-sambamba | Do not use sambamba for sorting SAM file, even when it is available. Use pysam instead. |
Mandatory arguments¶
fanc auto <input 1> <input 2> <input 3> < ... > <output folder>
fanc auto accepts any number of input files, which will be discussed below.
The last positional argument (without ‘-’) must always be the output folder for
all intermediate and final FAN-C files. fanc auto will generate the following
folder structure in the output folder:
output_folder
├── fastq
├── sam
├── pairs
├── hic
│ └── binned
└── plots
└── stats
You will find the processed files in these folders after completion of the pipeline - the folder names should be self-explanatory.
General arguments¶
fanc auto will name its output files using the -n parameter as prefix. If this is not
provided, it will try to come up with a basename from the largest overlap of the input file
names. As this can be surprising it is often best to specify the -n parameter directly.
You should also use the -t parameter to set the maximum number of parallel threads used
by fanc auto - by default, it uses only a single processor. It is highly recommended to
set this to a much higher value to enable parallel processing of large datasets.
If you are working on a network, or with multiple hard drives, you may want to use the -tmp
option. It instructs fanc auto to perform as many calculations as possible in a temporary
directory.
Input types¶
For this tutorial, we are going to use the example data provided on our
GitHub page in the examples folder.
It is a downsampled Hi-C library of a previously published human adrenal tissue dataset
(SRR4271982 of GSM2322539)
that only contains chromosomes 18 and 19.
Now let us discuss the different input types fanc auto can handle.
FASTQ input¶
To process FASTQ files with fanc, you must first have
Bowtie2 or
BWA installed on your system and available in your PATH.
Additionally, you need the corresponding index for the reference genome of your choice. We currently
recommend using BWA, as it supports chimeric reads, which are frequent in Hi-C libraries when
ligation junctions are sequenced.
Once you have these prerequisites, you can call fanc auto like this, assuming you are in the examples folder:
fanc auto SRR4271982_chr18_19_1.fastq.gzip SRR4271982_chr18_19_2.fastq.gzip \
./example_output/ -i bwa-index/hg19_chr18_19.fa \
-g hg19_chr18_19_re_fragments.bed
The first two arguments are the paired-end FASTQ files. fanc auto works with FASTQ and gzipped
FASTQ files. In general, fanc auto assumes that two consecutive FASTQ file arguments are mate
pairs (there is no pattern matching on _1 and _2 involved, so make sure you have the correct order
of input files!).
Note
When downloading FASTQ files from SRA using SRAtools, e.g. with fastq-dump, do not
use the -I / --readids option, which appends .1 or .2 to the read name. This
interferes with the sorting and read pairing step in FAN-C. Read names of the two mates
must be identical.
Following the FASTQ files as the last positional argument is the output folder
(example_output). -i or --genome-index instructs fanc auto to use the specified index
for mapping the FASTQ files to a reference genome. It will automatically determine whether a
BWA mem or Bowtie2 index is provided and choose the mapping software accordingly. Other mappers are
currently not supported (raise an issue on GitHub
if you are interested in support for your favourite mapper).
Warning
Always use the most comprehensive assembly of your genome of interest. Do not generate
and index from a subset of chromosomes. If you want to limit the chromosomes in your
Hi-C analysis, for example to canonical chromosomes, please do that using the -g
argument!
The last parameter (-g) is necessary for generating a fragment-level Hi-C map later in the
pipeline. This will be explained in more detail in the next section.
There are a few additional parameters that you can use to control the mapping process. Use
--iterative to iterative mapping: Reads are initially trimmed to 25bp before mapping, and
then iteratively expanded until a unique, high quality mapping location can be found. This can
improve mapping efficiency by a few percent, as smaller reads have a lower likelihood of mismatches
due to sequencing errors. -s or --step-size controls the size by which reads are extended
at every iterative mapping step.
As mentioned above, it is common to find reads in Hi-C libraries that contain a ligation junction
sequence. FAN-C can automatically split these kinds of reads before mapping using the
--split-ligation-junction option, which can improve mapping efficiency.
fanc auto parallelises mapping by spawning multiple mapping
processes internally. This can result in high disk I/O - if you have issues with poor performance,
try using the --mapper-parallel option, which will instead use the multithreading of your chosen
mapping software. If you are using Bowtie2, you can additionally use the --memory-map option,
which will load the entire Bowtie2 index into memory to be shared across all Bowtie2 processes. Use
this option if your system has a lot of memory available to speed up the mapping. Finally, if you
are using the -tmp option, which causes fanc auto to perform most pipeline steps in a
temporary directory, you may want to use the --split-fastq option to split the FASTQ files into
smaller chunks before mapping, so you can save space on your tmp partition.
The resulting BAM files are automatically handed to the next step in the pipeline, or you can
provide SAM/BAM files to fanc auto directly. This is described in the following section.
You can also perform the mapping separately with the fanc map command, which also gives you
additional options for controlling the mapping process, and which is described in
fanc map: Mapping FASTQ files.
SAM/BAM input¶
To process SAM/BAM files, no additional external software is required. However, we do recommend the installation of Sambamba, which can greatly speed up the SAM sorting step required for merging mate pairs into the Pairs object.
A minimal fanc auto command using SAM/BAM files could look like this:
fanc auto output/sam/SRR4271982_chr18_19_1.bam output/sam/SRR4271982_chr18_19_2.bam \
./example_output/ -g hg19_chr18_19_re_fragments.bed
Similarly to FASTQ input, fanc auto assumes that two consecutive SAM/BAM files represent
mate pairs, and will match the read names in the pairing step. The -g or --genome
parameter is mandatory for both FASTQ and SAM/BAM input, and is used to load (or construct) the
restriction fragment regions necessary for building the fragment-level Hi-C object.
You can either directly provide a region-based file with restriction fragments (most file
formats are supported, including BED and GFF), or use a FASTA file with the genomic sequence
in conjunction with the -r or --restriction-enzyme parameter. In the latter case,
fanc auto will perform an in silico digestion of the genome and use the resulting
restriction fragments from there.
Note
Genome assembly FASTA files typically contain a large number of unassembled contigs or
other sequences that are often irrelevant for downstream Hi-C analysis. As the number of
chromosomes can negatively affect FAN-C performance, it is generally a good idea to limit
the analysis to canonical chromosomes. A very easy way to do with with fanc is the
fanc fragments command, which accepts a --chromosomes option to specify exactly
which chromosomes you want in the final analysis. The output file can be directly used as
input for the -g argument.
SAM/BAM files are first sorted and then matched by qname. Together with the restriction
fragment list, mate pairs will be assigned to restriction fragments and stored in a “Pairs”
object. By default, fanc auto excludes unmappable and multimapping reads, as these are
unusable or misleading in interpreting Hi-C data. Additional filters for read pairs are
described in the Pairs input section.
You can run the SAM/BAM to Pairs step of the fanc auto pipeline separately using
fanc pairs, which is described in mor detail in fanc pairs: Generating and filtering read Pairs.
“Valid pairs” txt input¶
Many tools for processing Hi-C data output “valid pairs” files, which are typically tab-delimited text files that contain read pair information. FAN-C supports valid pairs files from HiC-Pro and the 4D Nucleome project.
With fanc auto you can load them like this
fanc auto test.validPairs ./example_output/ -g hg19_chr18_19_re_fragments.bed
fanc auto will attempt to automatically determine if you supply a valid pairs file.
Pairs input¶
If you already have a FAN-C Pairs object, for example from a previous fanc auto run or
from the fanc pairs command, you can feed them to fanc auto directly:
fanc auto output/pairs/fanc_example.pairs ./example_output/
The Pairs objects already contain restriction fragment information, hence the -g parameter
is no longer necessary. Unless using the --no-filter-pairs option, fanc auto will first
filter read pairs for self-ligated fragments, PCR duplicates, and restriction site distance
(>10kb). You have the option to additionally filter out ligation error products using the
--le-inward-cutoff and --le-outward-cutoff parameters. More details on the different
filtering options are available in the description of the separate fanc pairs command:
fanc pairs: Generating and filtering read Pairs
After filtering, Pairs files are converted to fragment-level Hic objects. The parameters applying to their processing are described in the next section.
Hic input¶
If you already have a FAN-C Hic object, for example from a previous fanc auto run or
from the fanc hic command, you can feed them to fanc auto directly:
fanc auto output/hic/fanc_example.hic ./example_output/
If you are running this command with multiple input files, these will be merged into a single
fragment-level Hic object. This merged Hic object will then be binned at the resolutions
specified with the -b parameter. By default, it will produce binned Hic files at 5mb,
2mb, 1mb, 500kb, 250kb, 100kb, 50kb, 25kb, 10kb, and 5kb resolution.
Binned Hi-C files will be filtered for coverage (bins with less reads than 10% of the median
bin coverage) and corrected using Knight-Ruiz matrix balancing. If you prefer ICE correction,
use the --norm-method ice option, or --norm-method vc for vanilla coverage normalisation.
Each chromosome in the matrix is corrected independently, and
by default the corrected matrix entries correspond to contact probabilities. You can use the
--restore-coverage option to force matrix entries in a chromosome to sum up to the
total number of reads before correction.
You can run the Hi-C processing step independently with the fanc hic command, as described
in detail in fanc hic: Generating, binning, and filtering Hic objects
Mixed input¶
Now that we have covered all the different input options fort fanc auto, it is worth
stressing that you can combined different types of input in the same command. fanc auto
will attempt to automatically determine the commands necessary for each input to run
through the entire pipeline, and will merge inputs into a single fragment-level Hic object
before binning.
That means something like this is possible (although it does not make sense in this particular case):
fanc auto hic/test.hic pairs/test.pairs test.validPairs \
sam/SRR4271982_chr18_19_1.bam sam/SRR4271982_chr18_19_2.bam \
SRR4271982_chr18_19_1.fastq.gzip SRR4271982_chr18_19_2.fastq.gzip \
./example_output/ -g hg19_chr18_19_re_fragments.bed -b 1mb, 50kb, 25kb \
-i bwa-index/hg19_chr18_19.fa -n test -s 20 -t 16 -q 3
Test runs and Sun/Oracle/Slurm Grid engine support¶
By default, fanc auto runs tasks in parallel locally on the machine it was started on.
If you want to perform a test run, without actually executing any commands, you can use
the --run-with test option. This will not run any of the fanc pipeline steps, but
will print each command it would run, including the dependencies between commands, to the
command line.
If you have access to a computational cluster running Sun/Oracle Grid Engine (SGE/OGE), you
can instruct fanc auto to submit all commands to the cluster using --run-with sge.
Internally, this calls qsub on each command and uses the --hold_jid parameter to
ensure each command waits for the output of its dependencies. You can configure the SGE
setup using FAN-C config files
There is also experimental support for Slurm, which you can
enable using --run-with slurm.
Next steps¶
Once you have generated your binned, filtered, and corrected Hic objects with fanc auto,
you may want to explore the data in those matrices. FAN-C provides a number of commands for
data analysis and exploration. Continue with Analyse Hi-C matrices (API) for further details.
If you want to explore individual matrix generation pipeline steps, continue to Individual pipeline steps.